
High-Throughput Epigenomics
ATAC-seq
Assay for Transposase-Accessible Chromatin coupled with high-throughput sequencing
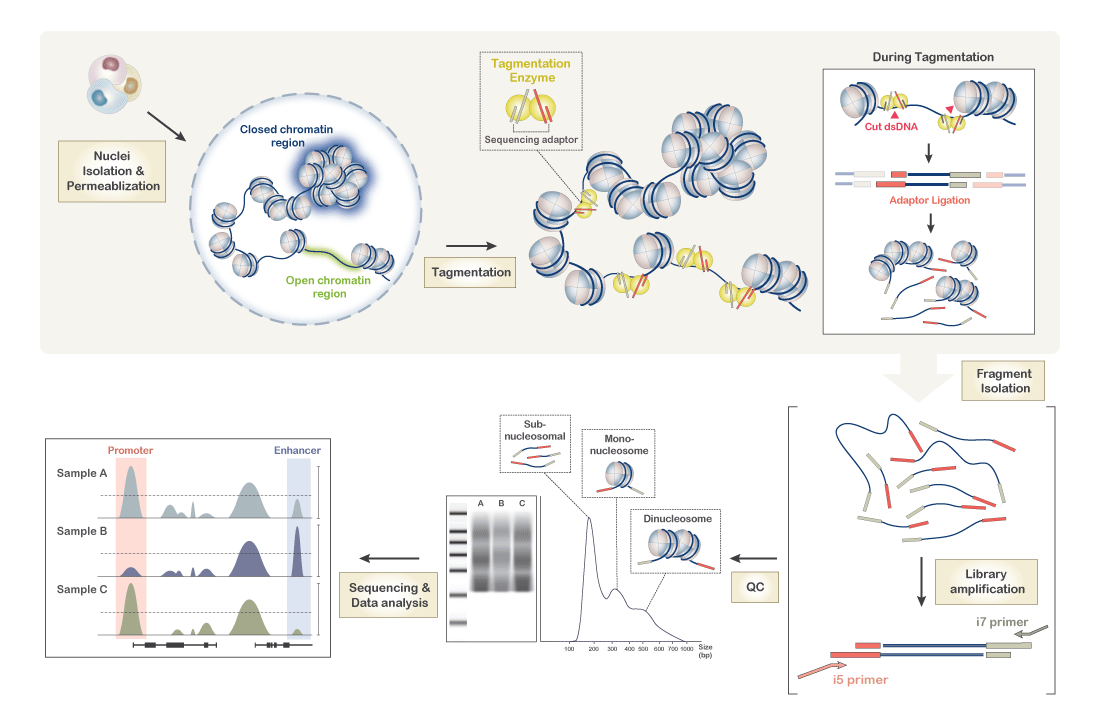
How is ATAC-seq useful for my research?
ATAC-seq identifies regions of the genome that have the potential for regulatory activity. These regulatory regions are accessible to the binding of regulatory proteins that influence gene expression, and, thus, also accessible to the transposase enzyme used in ATAC-seq. Identifying regulatory regions can provide valuable insights into the transcriptional regulators and signaling pathways active in a cell type or tissue of interest. For more information please see the following publications:
Buenrostro JD et al. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015 Jan 5;109:21.29.1-9.
Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017 Oct;14(10):959-962.
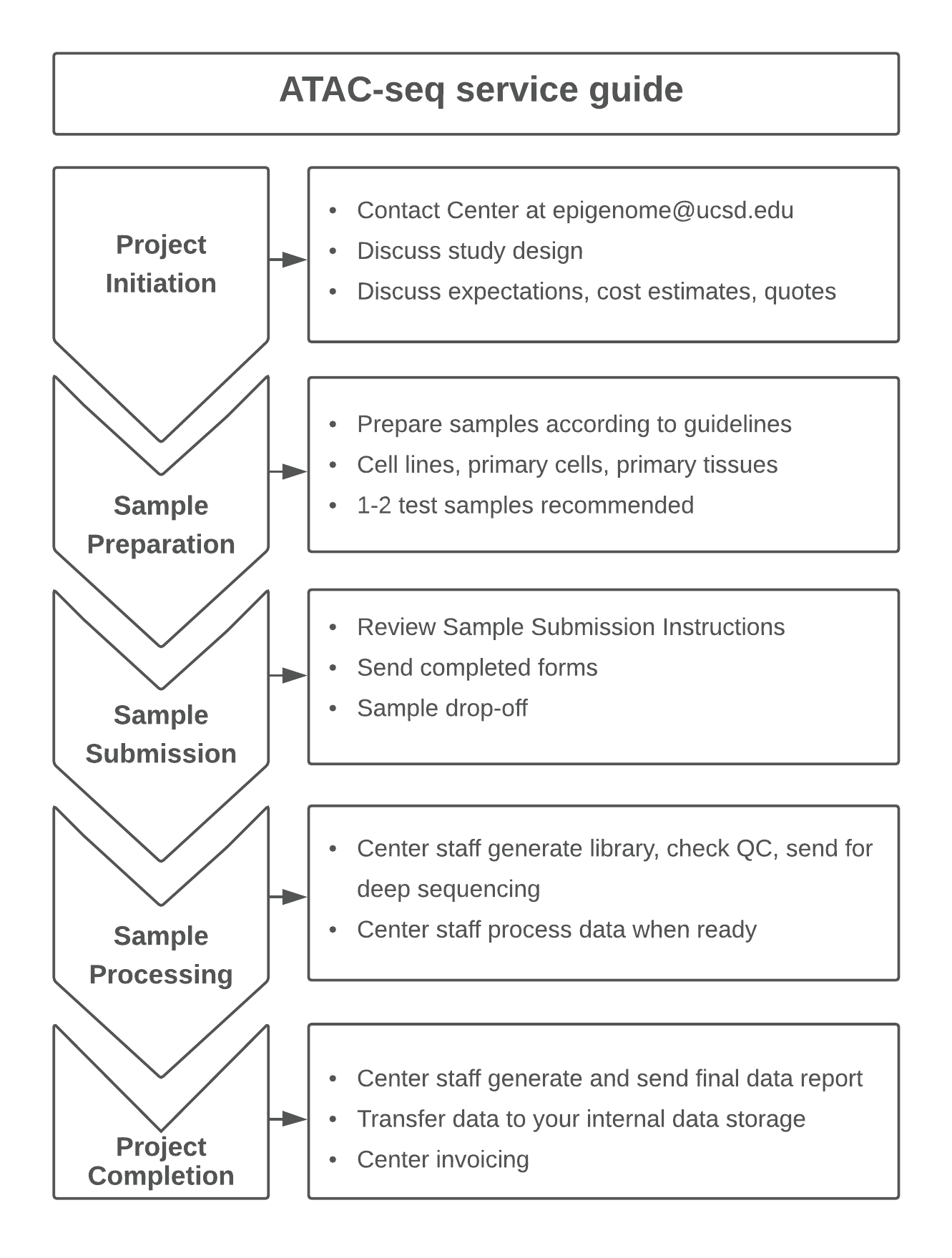
How do I initiate an ATAC-seq project with the Center for Epigenomics?
Typical ATAC-seq projects follow the workflow below which ensures your samples are prepared, submitted, and processed appropriately.
If you are interested in our ATAC-seq Service, please contact us at epigenome@ucsd.edu to discuss your ideas.
Looking for specific information about ATAC-seq with the Center?
- Sample submission instructions for UC San Diego Investigators (PDF)
- Sample submission instructions for Non-UC San Diego Investigators (PDF)
- Sample preparation guide (PDF)
- Sample drop-off map (PDF)
- Example final data report
- Final data report handbook
- ATAC-seq protocol used for cells (PDF) or tissues (PDF)
FAQs
Project Initiation
What is included in the ATAC-seq service?
The ATAC-seq service includes nuclei preparation, library preparation, sequencing, and standard data processing. A final data report (example) will be provided upon completion of the service. To review further details of this service see ATAC-seq content above.
What is the cost of ATAC-seq per sample?
The cost of ATAC-seq service can be broken down into two parts:
- Library preparation: This includes all reagents and labor costs associated with library preparation. Basic data processing and quality analysis are also included in this portion of the cost.
- Sequencing: Our deep sequencing is performed at the UCSD IGM Genomic Center at their standard rates. We do not charge additional fees for sequencing. Unless otherwise agreed upon, we target ≥25M usable read pairs per library as per ENCODE standards.
To obtain more specific cost estimates for your samples, contact the Center at epigenome@ucsd.edu.
How do I initiate a new ATAC-seq project?
To initiate a new project, please contact the Center at epigenome@ucsd.edu. After discussing the project, we will provide detailed instructions for sample preparation and drop-off.
Sample Preparation
What kind of samples are recommended for ATAC-seq, and how should they be prepared?
ATAC-seq can be performed on cell lines, primary cells, and primary tissue. See our preparation guide for sample preparation guidelines. For new customers or new sample types, we strongly recommend that 1-2 test samples be submitted first before moving forward with a larger set of samples.
What if my sample type is not included in your preparation guide?
Contact epigenome@ucsd.edu if your samples do not conform to the sample preparation guidelines.
My samples have been frozen for years, can you still process them?
If samples are frozen according to guidelines they can remain stable for extended periods of time at -80°C. In some cases, we have observed that sample quality can drop over time. The best way to determine the answer for your samples is to test them in our workflow.
Sample Submission
Where can I drop-off my samples?
Please make sure that you have contacted epigenome@ucsd.edu, and that we are expecting your samples before you drop them off. If you are a UCSD investigator, or local to the La Jolla/San Diego area, you may drop off your samples following these instructions for UCSD (PDF). Alternatively, you can ship your samples to us (see below).
Can I ship my samples to you?
Please make sure that you have contacted epigenome@ucsd.edu, and that we are expecting your samples before you ship them. You may ship samples to our Center following these instructions for non-UCSD (PDF).
Sample Processing
How long will it take to process my samples?
Samples are queued in the order in which they are received (with completed paperwork). The queue and processing times vary, but on average it takes 6-8 weeks from sample submission to final data delivery. This timeline is only a guideline and is not guaranteed.
What protocol is used for ATAC-seq service?
Our ATAC-seq protocol is adapted with modifications from Buenrostro et al., 2015 (PMID: 25559105). Detailed protocols: for cells (PDF) and for tissue (PDF). An example paragraph suitable for publication of methods is available: example (DOCX). Please note that buffer compositions may be adjusted for certain sample types based on our previous experience. Please confirm protocol details with the Center prior to publication (epigenome@ucsd.edu).
What kind of data will I receive from this service?
Each sample submitted will result in one ATAC-seq dataset. The ATAC-seq data will be provided in the form of raw and processed data files, to include: "fastq" files containing the raw sequencing results, "bam" files containing alignments to a reference genome, and "bed" files with lists of regions called as enrichment peaks. The final data report will also include QC metrics and an integrated genome browser for manual exploration of the data.
How was my ATAC-seq data processed?
Documentation and code for our ATAC-seq data processing pipeline is available on GitHub. It is based on the Kundaje lab's pipeline designed for ENCODE.
How do I analyze my ATAC-seq data?
In most cases, you will want to perform downstream analysis of the ATAC-seq data once you receive your assay results. These analyses require time and bioinformatic expertise. The Center can give advice on best practices, but, unfortunately, does not have the bandwidth to perform downstream analysis in most cases. For additional bioinformatics support, we recommend contacting the UCSD Center for Computational Biology & Bioinformatics which can provide ATAC-seq analysis services.
How do you determine how many sequencing reads are needed?
We aim for ≥25M usable read pairs after filtering for unmapped reads, multi-mappers, PCR duplicates, and mitochondrial reads. This number is a guideline based on standards set by the ENCODE consortium. To achieve this, we sequence each library on ~1/6th of a HiSeq4000 lane. Because read filtering rates vary by sample, we cannot guarantee the final number of usable reads.
Project Completion
How will I receive my sequencing data?
When sequencing and data processing are finished, you will receive an email with your final data report. This report contains quality metrics, an integrated genome browser, and instructions to download data files. We recommend downloading all files to your local storage as soon possible once the final report is received. We will host final report data on our servers for at least six months, but data is periodically deleted from our servers to make room for new files.
When will I receive an invoice for this service?
We consider your service complete once the final data report has been sent. In most cases, an invoice will be generated and sent to you within approximately 30 days of completion. For UC San Diego investigators, the provided index will be charged at this time. For external investigators, we ask that you forward payment within 30 days of receipt. In some cases, it may be necessary to arrange for pre-payments.
Other Questions
Does the Center perform library preparation only?
We only offer the full ATAC-seq service starting from cell or tissue samples. We do not offer standalone library preparation services.
What sort of quality control is performed?
Prior to deep sequencing, all ATAC-seq libraries undergo quaintly control (QC) analysis by shallow sequencing (~1-5M reads per library) to evaluate mapping rates, signal-to-noise ratio by Transcription Start Site (TSS) enrichment, and screen for sequence contamination. If abnormalities are observed at this time you will be contacted by Center staff with recommendations about whether to proceed with deep sequencing. A meeting can also be arranged with Center staff at this time to review QC data, though this is not always necessary.
What happens if my samples fail?
The quality of ATAC-seq data is highly dependent upon factors related to sample derivation and preparation which are out of the Center's control. We strongly recommend submitting 1-2 test samples of the same type and using the same preparation method to test data quality before submitting a larger batch of samples. As described above, all libraries will undergo QC by shallow sequencing prior to deep sequencing. If samples are terminated at this stage due to failed QC, you will be charged only for the library preparation, but not for deep sequencing. We include a positive control sample in each batch of ATAC-seq to monitor general integrity of reagents and workflow.
Do I own the data that result from this service?
Yes, you own the data from this service.
May I speak to someone about this service?
Yes, please contact the Center at epigenome@ucsed.edu to inquire about our fee for service operation.
What is the status of my samples?
You may inquire at epigenome@ucsd.edu about the status of your samples. As time permits, we may also periodically send a sample update to you.