
Single Cell Epigenomics Services
Our automated technology platforms enable cost-effective workflows to incorporate epigenomics into a variety of research projects. In some cases, we can offer epigenomic assays on a fee-for-service basis. Please see below for the single cell services available at the Center on a fee-for-service basis.
Single Cell Services
The Center for Epigenomics has 3 single cell genomics assays available for Epigenomic Services: sci-ATAC-seq, 10x-ATAC-seq, and 10x-sc/snRNA-seq. Review each assay's unique characteristics in the details below to determine which assay best fits your research needs.
sci-ATAC-seq
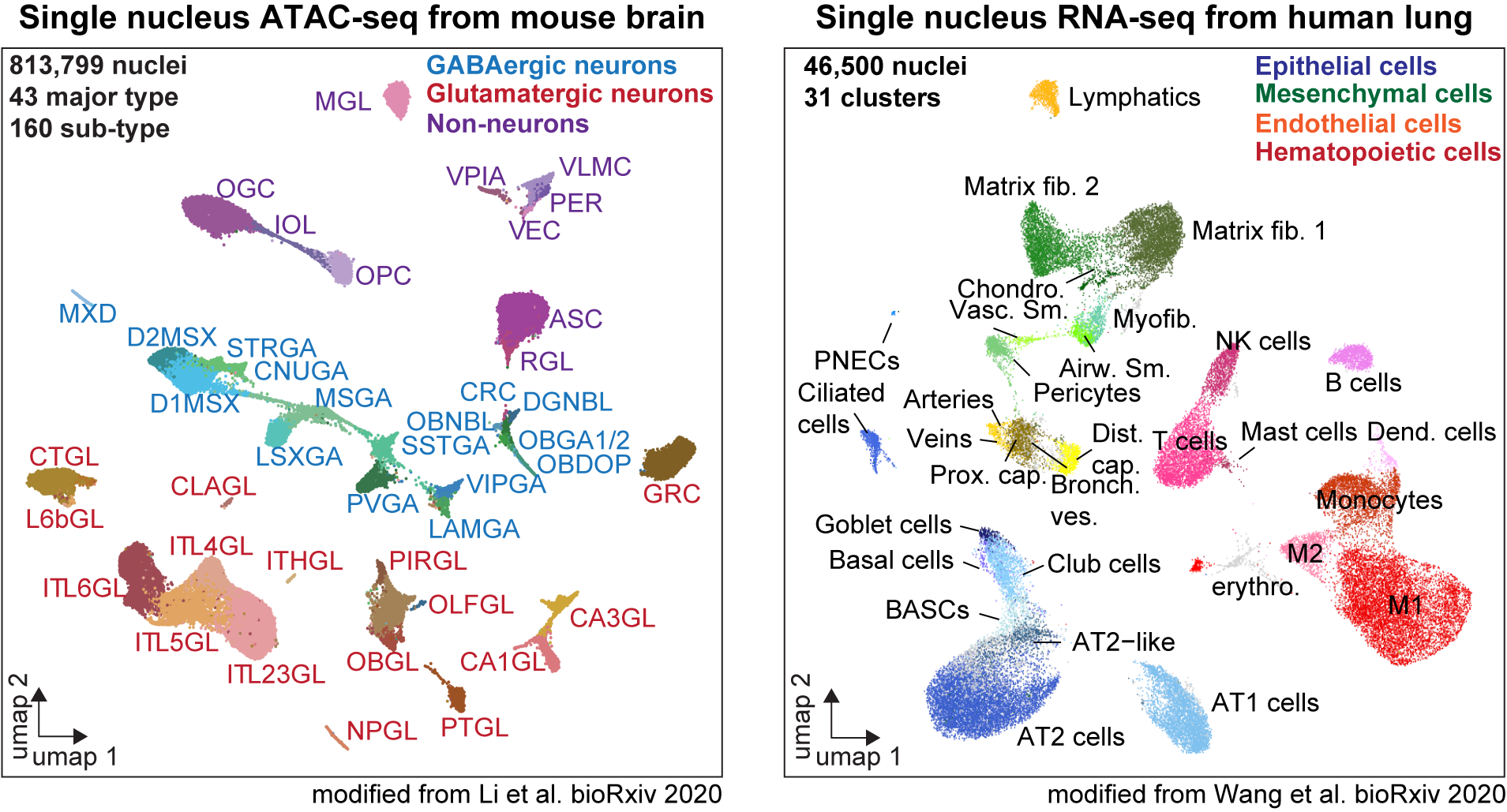
sci-ATAC-seq (single cell combinatorial indexing) originally developed by Jay Shendure's lab (Cusanovich et al. 2015) to resolve cellular heterogeneity and delineate transcriptional regulatory sequences in the constituent cell types. This platform offers high flexibility e.g. the ability to mix multiple samples in one run as well as the opportunity to run a small-scale experiment to identify best conditions or to evaluate sample quality. We usually capture ~10k nuclei per 96 well plate (these can be split between samples and/or conditions). After completion of the assay we will provide a data report based on our processing pipeline.
Example publications:
Human lung tissue
Mouse brain
Mouse mammary gland cells
Whole mouse embryo
Pancreatic islets
10x-ATAC-seq
A droplet based single cell ATAC-seq platform from 10x Genomics (Satpathy et al. 2019) used to resolve cellular heterogeneity and delineate transcriptional regulatory sequences in the constituent cell types. This platform works particularly well for cell lines, tissue with minimal debris, and samples with a low number of starting cells/nuclei. After the completion of the assay we will provide a standard Cell Ranger data report for each sample.
Example publications:
Please check back later for example publication
10x-sc/snRNA-seq
We use the droplet-based 10x Genomics Chromium Platform to generate 3'RNA expression profiles of thousands of cells or nuclei (Zheng et al. 2017). For other RNA expression profiling solutions, e.g. 5' profiling please contact us at epigenome@ucsd.edu. After completion of the assay we will provide a standard Cell Ranger data report for each sample.
Example publications:
Brain organoids
Human Lung
How do I initiate a single cell services project with the Center for Epigenomics?
All samples submitted for our single cell assays follow a typical workflow:

If you are interested in our single cell Services, please contact us at epigenome@ucsd.edu to discuss your ideas.
Looking for specific information about single cell assays with the Center?
- Sample submission instructions UCSD investigator (PDF)
- Sample submission instructions Non-UCSD investigator (PDF)
- Sample preparation guide sci- or 10x-ATAC-seq (PDF)
- Sample preparation guide 10x-RNA-seq (PDF)
FAQs
Project Generation
What services are you offering?
The Center for Epigenomics has 3 single cell assays available for Epigenomics Services:
- Single cell/nucleus ATAC-seq using our in-house semi-automated combinatorial barcoding platform (sci-ATAC-seq)
- Single cell/nucleus ATAC-seq using the Chromium Single Cell ATAC Library v1.1 (10x-ATAC-seq; 10x Genomics)
- Single cell/nucleus RNA-seq using the Chromium Single Cell 3ʹ v3 solution (10x-sc/snRNA-seq, 10x Genomics)
What is included in each single cell service?
The Single Cell Genomics service includes sample preparation, library preparation, sequencing, and standard initial data processing using in-house pipelines and/or Cell Ranger (10x Genomics).
What costs are associated with single cell assays?
The cost of Single Cell Genomics services can be broken as follows:
- Library preparation and initial data processing: This includes all reagents and operational costs associated with sample preparation and library preparation. Operational costs are included in the cost for the first sample of each batch. For additional samples we will charge only reagent costs and technician hours required for processing larger batches. For sci-ATAC-seq, batches up to 384 (4 x 96 well) tagmentation reactions are possible. For 10x-ATAC-seq and 10x-sc/snRNA-seq we recommend batches of 4-8 depending on requirements for sample preparation. Quality control analysis and initial data processing are included in this portion of the cost.
- Sequencing: Shallow sequencing for quality control of the libraries is usually performed on a NextSeq500 in the Center for Epigenomics and costs consists of reagents and operational costs. Deep sequencing is usually performed at the UCSD IGM Genomic Center at their standard rates. We do not charge additional fees for sequencing.
- Pilot experiments: Mimic sci-ATAC-seq experiments will be charged based on reagents and technician hours.
To obtain more specific cost estimates for your samples, please contact the Center at epigenome@ucsd.edu.
How do I initiate a new single cell project?
To initiate a new project, please contact the Center at epigenome@ucsd.edu. After discussing the project, we will provide detailed instructions for sample preparation and drop-off.
What is the difference between 10x-ATAC-seq and sci-ATAC-seq?
- sci-ATAC-seq offers high flexibility e.g. the ability to mix multiple samples in one run as well as the opportunity to run a small-scale experiment to identify best conditions or to evaluate sample quality. We usually capture ~10k nuclei per 96 well plate (these can be split between samples and/or conditions).
- 10x-ATAC-seq works particularly well for cell lines, tissue with minimal debris and samples with a low number of starting cells/nuclei. Nuclei sorting is an option for some tissues prior to 10x-ATAC-seq. We will use 15,300 cells/nuclei as input for 10x-ATACseq library preparation. We usually recover ≥5,000-12,000 profiles per sample. These libraries typically yield 5-10 fold more fragments/nucleus but lower TSS (transcriptional start site) enrichment as compared to sci-ATAC-seq. Recovery rate varies between sample types.
What kind of pilot experiments would you recommend for my samples?
The Center for Epigenomics recommends 1-2 test samples be submitted as a pilot experiment to test your samples in our workflow.
For sci-ATAC-seq- the pilot experiments depend on our previous experience with the sample and your sample amount:- If it is a new sample type, we recommend a preliminary sci-ATAC-seq mimic experiment to test the nuclei preparation method (tissue homogenization and nuclei permeabilization). We will use qPCR to estimate library complexity and signal-to-noise ratio. We will also save an aliquot for potential sequencing.
- If we have experience with the sample type, we recommend a sci-ATAC-seq test run with a few samples on a split 96-well plate for tagmentation. We will use a small aliquot of your sample to perform a small-scale sci-ATAC-seq run to evaluate sample quality. These libraries will be shallow sequenced, and therefore, provide small scale data for preliminary analysis.
- After completion of pilot experiments, we will provide a report summarizing the best processing method, pictures of nuclei suspension, extracted nuclei per milligram of tissue, signal-to-noise ratio or TSSe and an estimation of library complexity.
- If it is a new sample type, we will establish best practices for sample handling using a Bulk ATAC-seq experiment to determine nuclei preparation method (tissue homogenization and nuclei permeabilization) This library will be shallow sequenced to determine transcriptional start site (TSS) enrichment. Please note: We have seen samples perform well in bulk ATAC-seq but fail in the 10x-ATAC-seq workflow. Therefore, this test is just to explore if the samples are in general amenable for ATAC-seq.
- If we have experience with the sample type, we recommend a test run with 1-2 samples or conditions following the 10x-ATAC-seq workflow to determine the samples behavior on the 10x Chromium controller, as well as expected nuclei recovery rate. We will use a small aliquot of your sample to perform a 10x ATAC-seq run loading a single well on the Chromium Chip. This library will be shallow sequenced for quality control (including TSS enrichment and estimated nuclei recovery rate), might be used for preliminary analysis (e.g. initial clustering) and could potentially be used for deeper sequencing.
- After completion of pilot experiments, we will provide a report summarizing the best processing method, pictures of nuclei suspension, extracted nuclei per milligram of tissue, signal-to-noise ratio or TSS enrichment and an estimation of library complexity. We will also provide standard Cell Ranger outputs.
Sample Preparation
What kind of samples are recommended for single cell assays, and how should they be prepared?
Single cell/nucleus ATAC-seq and single cell/nucleus RNA-seq can be performed on cell lines, primary cells, and primary tissue. Please see our guidelines for sample preparation (scATAC-seq and scRNA-seq). For new customers or new sample types, we strongly recommend that 1-2 test samples be submitted first before moving forward with a larger set of samples.
What is the required input amount for each assay?
- sci-ATAC-seq: ≥1M cells or 500k nuclei
- 10x-ATAC-seq (10x Genomics): >250k cells or nuclei
- 10x-scRNA-seq (10x Genomics): ≥250k cells
- 10x-snRNA-seq (10x Genomics): ≥250k nuclei
- 10x-sc/snRNA + sci/10x-ATAC-seq (from same sample): ≥500k cells or nuclei
Note: Numbers above are general guidelines. If you cannot fulfill them contact us at epigenome@ucsd.edu to discuss the best way forward for your samples.
What if my exact sample type is not included in your preparation guide?
Please contact epigenome@ucsd.edu if your samples do not conform to the sample preparation guidelines.
My samples have been frozen for years; can you still process them?
If samples are frozen according to our guidelines — see scATAC-seq (PDF) and scRNA-seq (PDF) — they can remain stable for extended periods of time at -80°C. In some cases, we have observed that sample quality can drop over time. The best way to determine the answer for your samples is to test them in our workflows.
Sample Submission
Where can I drop-off my samples?
Please make sure that you have contacted epigenome@ucsd.edu, and that we are expecting your samples before you drop them off. If you are a UCSD investigator, or local to the La Jolla/San Diego area, you may drop off your samples following these instructions for UCSD investigators (PDF). Alternatively, you can ship your samples to us (see below).
Can I ship my samples to you?
Please make sure that you have contacted epigenome@ucsd.edu, and that we are expecting your samples before you ship them. You may ship samples to our Center following these instructions for non-UCSD investigators (PDF).
Sample Processing
How long will it take to process my samples?
Samples are queued in the order in which they are received (with completed paperwork). The queue and processing times vary, but on average it takes 6-8 weeks from sample submission to initial data delivery (excluding deep sequencing). This timeline is only a guideline and is not guaranteed.
What protocol is used for single cell service?
For sci-ATAC-seq we use a custom protocol. For 10xATAC-seq and 10xsc/snRNA-seq (10x Genomics) we follow the user guide with minor modification based on experience. Please confirm protocol details with the Center prior to publication (epigenome@ucsd.edu). Detailed protocols can be found here. Please confirm protocol details with the Center prior to publication (epigenome@ucsd.edu).
How many single cell/nucleus profiles will I get?
Capture rates vary between samples and sample types; therefore, the below are just estimates.
- sci-ATAC-seq: sorted nuclei 15,360 --> targeting ~10k nuclei
- 10x-ATAC-seq: 15,300 loaded nuclei --> targeting ~10k nuclei (Note: depending on sample type we have seen as low as 3.5k nuclei)
- 10x-snRNA-seq: 12,000 loaded nuclei --> targeting ~8k nuclei
- 10x-scRNA-seq: 12,000 loaded cells --> targeting ~8k cells
How do you determine how many sequencing reads are needed?
The Center has determined the target number of sequencing reads based on our experience in combination with recommendations from 10x Genomics recommendations for 10x-ATAC-seq and 10xsc/snRNA-seq. Our established targets are:
Shallow sequencing for quality control:
- For 10x-ATAC-seq and 10x-sc/snRNA-seq we suggest shallow sequencing to assess library quality: 10-25M reads on a NextSeq 500/550 (Illumina).
Deep sequencing on a NovaSeq 6000 (Illumina) at the IGM CORE we use the following guidelines based on estimated target cell/nucleus numbers:
- sciATAC-seq: 150M paired-end reads (15k/nucleus)
- 10x-ATAC-seq: 250M paired-end reads (25k/nucleus)
- 10x-snRNA-seq: 160M paired-end reads (20k/nucleus)
- 10x-scRNA-seq: 200M paired-end reads (25k/cell)
Note: Deeper sequencing for 10x Genomics libraries is in many cases possible if budget permits.
What kind of data will I receive from this service?
Each sample submitted will result in one single cell/nucleus dataset. The data will be provided in the form of raw (FASTQ files) and processed data files. For sci-ATAC-seq data, we will provide files for mapped reads, data matrices, initial clustering and peak regions for individual clusters. Samples processed using the Chromium platform (10x Genomics) will include standard Cell Ranger outputs.
Which organisms may be submitted for single cell assays?
We accept samples from all organisms. Our standard processing pipelines use human (hg19, hg38), mouse (mm10), and rat (rn6) genomes. For other genomes we ask that you kindly provide a reference. Please note that several quality control metrics for ATAC-seq, e.g. transcriptional start site (TSS) enrichment, can vary between organisms and reference annotations used.
How was my single cell data processed?
For processing of sci-ATAC-seq data we use SnapATAC package with slight modifications. In addition, we use Cell Ranger for preprocessing of 10x-ATAC-seq and 10x-sc/snRNA-seq.
How do I analyze my single cell data?
In most cases, you will want to perform downstream analysis of the single cell/nucleus ATAC-seq and RNA-seq data once you receive your results. These analyses require time and bioinformatic expertise. The Center can give advice on best practices, but, unfortunately, does not have the bandwidth to perform downstream analysis in most cases. For additional bioinformatics support, we recommend contacting the UCSD Center for Computational Biology & Bioinformatics.
Project Completion
How will I receive my sequencing data?
When sequencing and data processing are finished, you will receive an email with your final data report. This report contains quality metrics, an integrated genome browser, and instructions to download data files. We recommend downloading all files to your local storage as soon possible once the final report is received. We will host final report data on our servers for at least six months, but data is periodically deleted from our servers to make room for new files.
When will I receive an invoice for this service?
We consider your service complete once the final data report has been sent. In most cases, an invoice will be generated and sent to you within approximately 30 days of completion. For UC San Diego investigators, the provided index will be charged at this time. For external investigators, we ask that you forward payment within 30 days of receipt of invoice. In some cases, it may be necessary to arrange for pre-payments.
How long do you store generated libraries after sequencing? Can I receive the library generated from my sample after sequencing?
Libraries are stored for up to a year, and we periodically clean our freezers to make room for new libraries. After completion of the work you may request transfer of the libraries.
Other Questions
Does the Center perform library preparation only?
We only offer the full Single Cell Genomics service starting from cell or tissue samples. We do not offer standalone library preparation services.
What sort of quality control is performed?
sci-ATAC-seq:
Before sequencing we evaluate nuclei suspension, extracted nuclei per milligram of tissue, nuclei sorting profile, fragment distribution, and library amount.
After sequencing, we evaluate transcriptional start site enrichment (TSSe), nuclei number passing quality control and fragments/nucleus.
10x-ATAC-seq:
We perform shallow sequencing and will provide a report summarizing pictures of nuclei suspension, extracted nuclei per milligram of tissue, sorting profile (if applicable) and standard Cell Ranger outputs including TSS enrichment and an estimation of library complexity.
10xsc/snRNA-seq:
We perform shallow sequencing and will provide a report summarizing pictures of nuclei/cell suspension, sorting profile (if applicable), and standard Cell Ranger outputs.
What happens if my samples fail?
The quality of single cell data is highly dependent upon factors related to sample derivation and preparation which are out of the Center's control. We strongly recommend submitting 1-2 test samples of the same type and using the same preparation method to test data quality before submitting a larger batch of samples. As described above, all libraries generated with the Chromium platform (10x Genomics) will undergo quality control (QC) by shallow sequencing prior to deep sequencing. If samples are terminated at this stage due to failed QC, you will be charged only for the library preparation and shallow sequencing, but not for deep sequencing.
Do I own the data that result from this service?
Yes, you own the data from this service.
What is your authorship policy?
Payment of fees for Center for Epigenomics services and authorship on publications, grants, and/or patents are not mutually exclusive. Depending on their contributions to a project, Center personnel should be considered collaborators at the same level as other academic colleagues who contribute intellectually and receive funding for work on a project. We follow the general guidelines for authorship laid out by the International Committee of Medical Journal Editors. The recovery of Center expenses through the recharge system does not exclude the possibility for authorship for Center personnel. Similarly, authorship does not substitute for payment of Center expenses for services rendered. If authorship is not appropriate, we kindly request that the Center for Epigenomics at UC San Diego is included in the acknowledgement section for our services with the statement that the appropriate assay "...was performed at the Center for Epigenomics at UC San Diego, which is partly funded by the UC San Diego School of Medicine." We also request that we are notified if data we generated is published.
May I speak to someone about this service?
Yes, contact the Center at epigenome@ucsd.edu to inquire about our Single Cell Services.
What is the status of my samples?
You may inquire at epigenome@ucsd.edu about the status of your samples. As time permits, we may also periodically send a sample update to you.